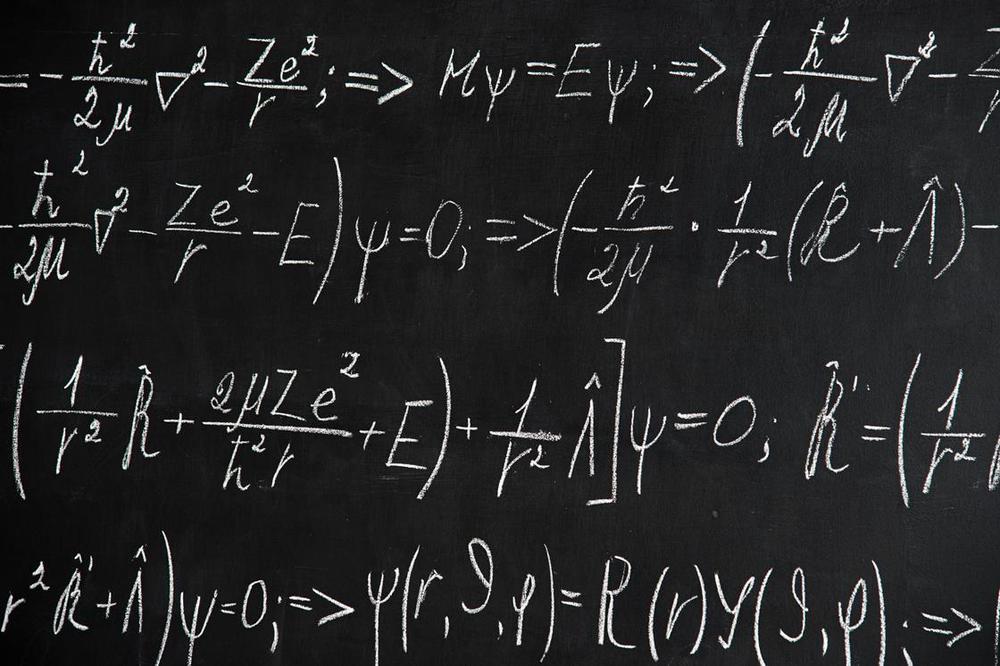
Using machine learning three groups, including researchers at IBM and DeepMind, have simulated atoms and small molecules more accurately than existing quantum chemistry methods. In separate papers on the arXiv preprint server the teams each use neural networks to represent wave functions of electrons that surround the molecules’ atoms. This wave function is the mathematical solution of the Schrödinger equation, which describes the probabilities of where electrons can be found around molecules. It offers the tantalising hope of ‘solving chemistry’ altogether, simulating reactions with complete accuracy. Normally that goal would require impractically large amounts of computing power. The new studies now offer a compromise of relatively high accuracy at a reasonable amount of processing power.
Each group only simulates simple systems, with ethene among the most complex, and they all emphasise that the approaches are at their very earliest stages. ‘If we’re able to understand how materials work at the most fundamental, atomic level, we could better design everything from photovoltaics to drug molecules,’ says James Spencer from DeepMind in London, UK. ‘While this work doesn’t achieve that quite yet, we think it’s a step in that direction.’
Two approaches appeared on arXiv just a few days apart in September 2019, both combining deep machine learning and Quantum Monte Carlo (QMC) methods. Researchers at DeepMind, part of the Alphabet group of companies that owns Google, and Imperial College London call theirs Fermi Net. They posted an updated preprint paper describing it in early March 2020.1 Frank Noé’s team at the Free University of Berlin, Germany, calls its approach, which directly incorporates physical knowledge about wave functions, PauliNet.2